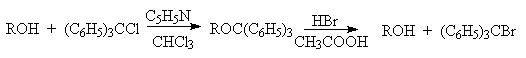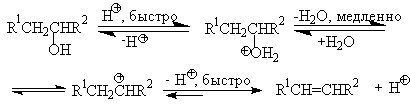
3.Дегидратация спиртов
Протонирование спиртов в ненуклеофильной среде приводит к дегидратации, котоая происходит при нагревании спирта в концентрированной серной, фосфорной кислотах или в суперкислой среде - смеси пятифтористой сурьмы и фторсульфоновой кислоты. Катион алкоксония, отщепляя воду, образует нестабильный интемедиат- карбокатион, который теряет протон с образованием алкена. Наиболее медленная стадия всего процесса - превращение катиона алкоксония в карбокатион. Концентрированная H2SO4 или H3PO4 связывают выделяющуюся воду, что делает весь процесс необратимым.
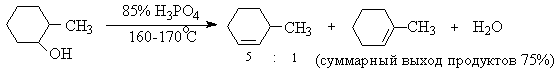
Приведенная последовательность превращений типична для реакций мономолекулярного элиминирования Е1, продукты которого определяются правилом Зайцева, т.е. преобладает наиболее разветвленный при двойной связи алкен. Вторичные спирты подвергаются дегидратациии при нагревании с 85%-ной
Фосфорной кислотой при 160-170оС или с 60-70%-ной серной кислотой при 90-100оС и направление дегидратации соответствует правилу Зайцева.
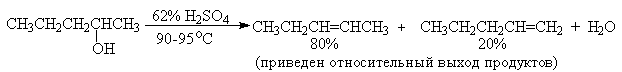
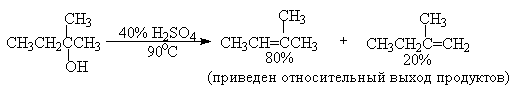
Дегидратацию третичных спиртов можно проводить уже в 20-50%-ной серной кислоте при 85-100oС.
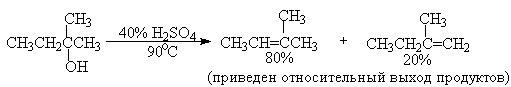
Третичные спирты дегидратируются так легко, что возможна избирательная дегидратация диола, содержащего третичную и первичную гидроксильные группы.
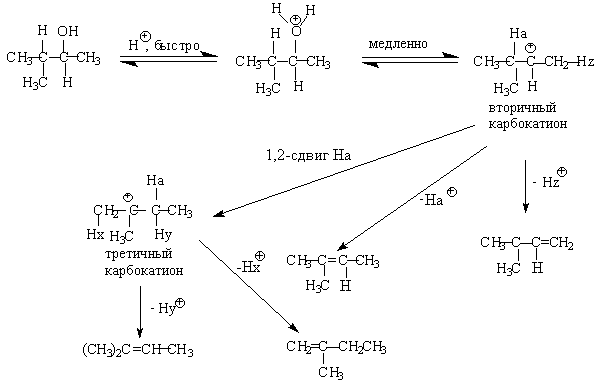
Для El-элиминирования, также как и для других процессов с образованием карбокатиона в качестве интермедиата, характерны перегруппировки, включающие анионотропную 1,2-миграцию гидрид-иона или алкильной группы. В качестве примера можно привести кислотно-катализируемую дегидратацию 3-метилбутанола-2 в 80%-ной серной кислоте.
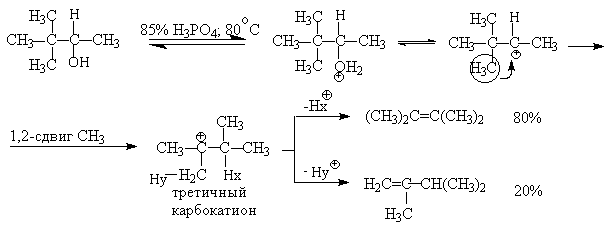
Примером скелетной изомеризации карбокатиона в Е1-элиминировании может служить дегидратация 3,3-диметилбутанола-2.
Для первичных спиртов, вероятно, реализуется иной, Е2 механизм дегидратации в концентрированной серной кислоте, они дегидратируются в гораздо более жестких условиях. Так, пропан ол-1 дает пропилен при нагревании с 96%-ной серной кислотой при 170-190oС, этанол в этих же условиях дает этилен.
![]()
Первичные спирты при взаимодействии с серной кислотой легко образуют полуэфиры серной кислоты. Е2-Элиминированию в этом случае, по-видимому, подвергается полуэфир, а роль основания выполняет гидросульфат-ион или вода.
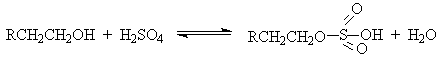
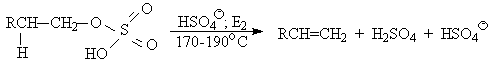
Возможен и другой механизм дегидратации первичных спиртов, в котором субстратом является катион алкоксония, а основанием гидросульфат-ион.
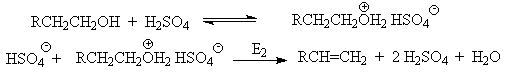
В более мягких условиях при нагревании простейших первичных спиртов с 96%-ной серной кислотой при 130-140oС преимущественно получаются простые эфиры. При этом первичный спирт алкилируется либо под действием полуэфира серной кислоты, либо при взаимодействии с катионом алкоксония.
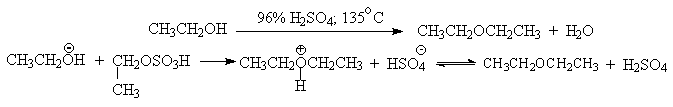
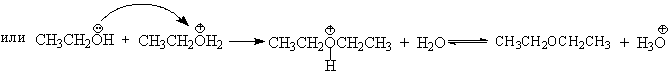
Этим способом получают простейшие простые эфиры - диэтиловый, дипропиловый и дибутиловый эфиры и циклические простые эфиры, например, тетрагидрофуран и диоксан. Вторичные и третичные спирты в этих условиях дегидратируются с образованием алкенов.
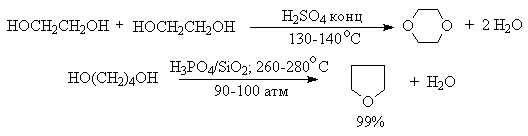
Этот способ неприемлем для получение несимметричных эфиров из двух спиртов, так как при этом образуется смесь трех возможных продуктов ROR, R'OR, R'OR'.
В промышленности для внутри- или межмолекулярной дегидратации вместо серной кислоты в качестве дегидратирующего средства используют безводную окись алюминия. Гетерогенная каталитическая дегидратация первичных, вторичных и третичных спиртов над окисью алюминия при 350-450oС приводит к алкенам.
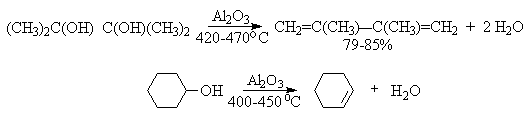
4. Получение простых эфиров по Вильямсону
Наиболее простым методом получения простых эфиров является реакция алкоголятов щелочных металлов с алкилгалогенидами или алкилсульфонатами (реакция Вильямсона, 1852 г.).
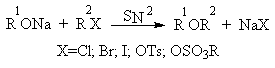
В отличие от межмолекулярной дегидратации спиртов реакция Вильямсона пригодна для синтеза как симметричных, так и несимметричных простых эфиров.
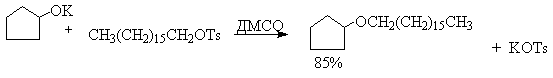


Получение простых эфиров по Вильямсону представляет собой обычную реакцию бимолекулярного нуклеофильного замещения у насыщенного атома углерода с помощью алкоксид- или феноксид-ионов. Если простой эфир содержит вторичную или третичную алкильные группы, ее следует вводить с помощью алкоголята, но не алкилгалогенида или алкилсульфоната, поскольку в противном случае преимущественно или исключительно будет происходить Е2-элиминирование.
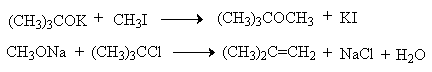
Наилучшие результаты достигаются в том случае, когда в качестве алкилирующего агента используются первичные алкил-, аллил- и бензилгалогениды и сульфонаты.
5.Окисление спиртов
Окисление первичных спиртов в альдегиды и вторичных спиртов в кетоны является одним из важнейших превращений функциональных групп и оценкой избирательного действия реагента, используемого в качестве окислителя.
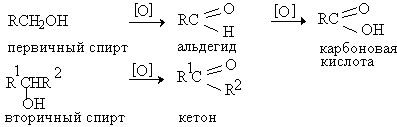
Третичные спирты не окисляются, а в жестких условиях окисление сопровождается деструкцией углеродного скелета. Наиболее широкое применение для окисления спиртов нашли реагенты на основе переходных металлов - хрома (VI), марганца (VII), марганца (IV).
Самостоятельную и наиболее сложную проблему при окислении первичных спиртов до альдегидов составляет дальнейшее окисление альдегидов до карбоновых кислот. Для предотвращения окисления альдегидов в карбоновые кислоты в качестве окислителя используют комплексы хромового ангидрида с третичными аминами, которые уменьшают окислительную способность окислителя и делают окисление более селективным. Лучшими реагентами для окисления первичных спиртов в альдегиды являются комплекс CrO3 с двумя молями пиридина (реагент Саррета-Коллинза) и хлорхромат пиридиния C5H5N+H.CrO3.Cl- (реагент Кори) в хлористом метилене. Реагент Саррета-Коллинза получается при медленном введении оксида хрома (VI) к пиридину при 10-15оС. Оранжевый комплекс CrO3 c пиридином и HСl получается при добавлении пиридина к раствору CrO3 в 20%-ной соляной кислоте. Оба эти реагента растворимы в CH2Cl2 или CHCl3.
Ниже приведены некоторые наиболее типичные примеры окисления первичных спиртов до альгедигов комплексами оксида хрома VI.
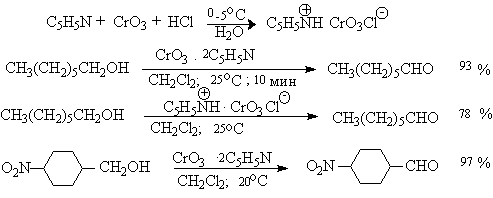
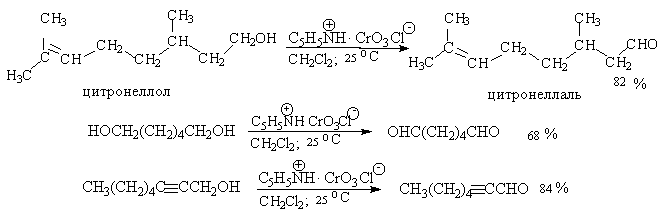
Оба окислителя обеспечивают очень высокие выходы альдегидов, однако хлорхромат пиридиния имеет важное преимущество, так как он не затрагивает двойную и тройную связи и может быть использован для получения ненасыщенных альдегидов.
Для получения a,b-ненасыщенных альдегидов окислением замещенных аллиловых спиртов универсальным окислителем является оксид марганца (IV) MnO2. Этот реагент окисляет в петролейном эфире или хлористом метилене ненасыщенные спирты с одной или несколькими двойными или тройными связями без изомеризации и перегруппировки, что с успехом используется в синтезе природных соединений.
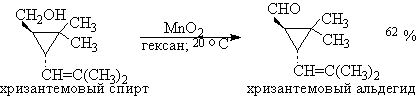
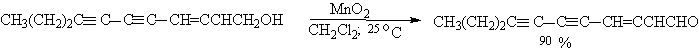
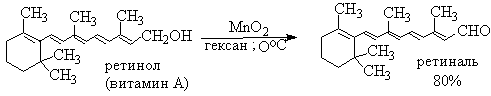
Комплексы хромового ангидрида с пиридином окисляют и вторичные спирты до кетонов с почти количественными выходами.
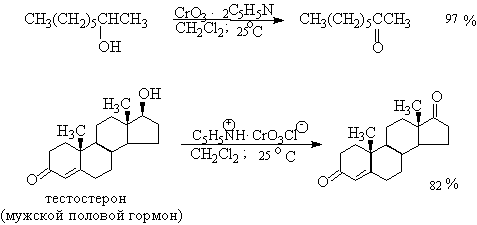
Однако чаще всего для окисления вторичных спиртов используют реактив Джонса - раствор строго рассчитанного количества CrO3 в водной серной кислоте. Важное достоинство реагента Джонса состоит в том, что вторичные спирты, содержащие двойную или тройную связь, быстро окисляются до кетонов без затрагивания кратных связей.
![]()
Первичные спирты окисляются реактивом Джонса до карбоновых кислот.
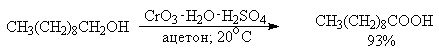
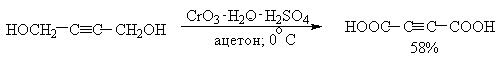
Механизм оксиления спиртов под действием хромового ангидрида подробно изучен. Эта реакция включает несколько стадий. Сначала из спирта и CrO3 образуется сложный эфир хромовой кислоты. Во второй, ключевой, стадии имеет место окислительно-восстановительное элиминирование, приводящее к образованию альдегида или кетона и частицы, содержащей Cr(IV). При окислении дейтерированного CH3CD(OH)CH3 и недейтерированного пропанола-2 наблюдается кинетический изотопный эффект КН/KD=7. Столь значительный первичный кинетический изотопный эффект показывает, что элиминирование является наиболее медленной стадией, определяющей скорость всего процесса.
![]()
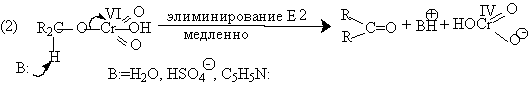
Установлено, что частицы, содержащие хром (IV), также принимают участие в окислении спирта.
Окисление под действием соединений Cr (IV) можно полностью подавить с помощью солей, содержащих ионы Mn (II) или Ce (III), которые окисляются Cr (IV). Каталитические количества Ce (IV) также подавляют эту побочную реакцию, поскольку Ce (IV) катализируют диспропорционирование Cr(IV) на Cr(III) и Cr (VI).
![]()
Для третичных спиртов, не содержащих атомов водорода при карбонильном углероде, эфиры хромовой кислоты могут быть выделены.
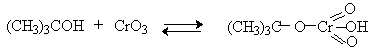
Раствор хромового ангидрида в трет-бутиловом спирте также используется для окисления первичных и вторичных спиртов. Раствор хромового ангидрида в уксусной кислоте нередко употребляется в качестве окислителя вторичных спиртов до кетонов.
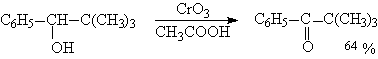
Механизм дальнейшего окисления альдегидов до карбоновых кислот по существу аналогичен механизму окисления спиртов. В водной среде альдегид находится в равновесии с геминальным 1,1-диолом, который образует сложный эфир с хромовым ангидридом. При элиминировании НCrO3- из этого сложного эфира получается карбоновая кислота.
![]()
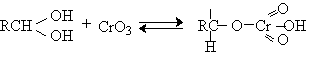
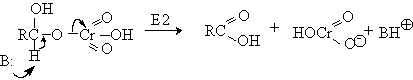
Поэтому для того, чтобы избежать дальнейшего окисления альдегида, окисление первичных спиртов следует проводить в апротонной среде при полном отсутствии влаги. Этому условию в полной мере удовлетворяют реагенты Коллинза и Кори, для которых в качестве растворителей используют тщательно обезвоженный хлористый метилен.
В последние тридцать лет разработано несколько эффективных способов окисления первичных и вторичных спиртов с помощью ДМСО или комплексов ДМСО с электрофильными агентами. Тозилаты первичных спиртов, также как и бензилтозилаты, окисляются в альдегиды при нагревании в ДМСО в течение 10-30 минут при 120-150оС в присутствии гидрокарбоната натрия как слабого основания.
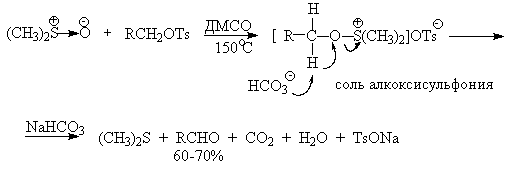
ДМСО в этой реакции выполняет роль нуклеофильного агента, который замещает тозилоксигруппу по обычному SN2 механизму с образованием алкоксисульфониевой соли. Катион алкоксисульфониевой соли далее подвергается окислительно-восстановительному элиминированию по механизму, аналогичному для окислительно-восстановительного элиминирования из сложных эфиров хромовой кислоты. Гидрокарбонат-ион является основанием в этой Е2 реакции элиминирования, приводящей к диметилсульфиду и альдегиду. В качестве примера приведем получение гептаналя и и-бромбензальдегида.

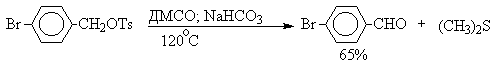
Слабый нуклеофильный агент ДМСО легко превращается в сильный электрофильный агент, который реагирует со спиртами уже ниже 0oС в мягких условиях. Необходимую активацию ДМСО проводят с помощью серного ангидрида, трифторуксусного ангидрида, N-хлорсукцинимиджа или N,N-дициклогексилкарбодиимида C6H11N=C=NC6H11 (ДЦГК). Во всех случаях в качестве реакционноспособного интермедиата образуется активированная алкоксисульфониевая соль, которая далее подвергается внутримолекулярной окислительно-восстановительной фрагментации.
Для окисления первичных и вторичных спиртов до альдегидов и кетонов в мягких условиях эффективен комплекс ДМСО с SOs, образующийся при взаимодействии пиридинсульфотриоксида с ДМСО.
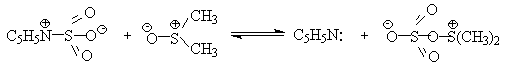
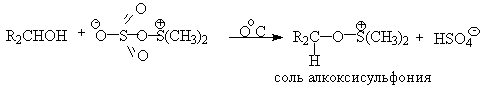
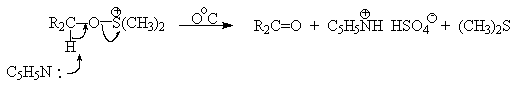
SO3 в качестве электрофильной частицы может быть заменен трифторуксусным ангидридом или ДЦГК (реактив Пфитцера-Моффата). Этот реагент в настоящее время употребляется наиболее часто.
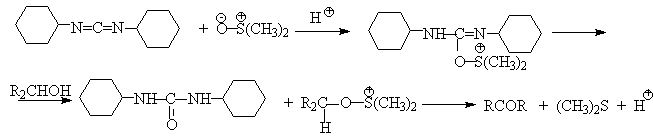
Приведем для иллюстрации два примера окисления спиртов комплексами ДМСО.
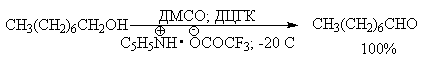
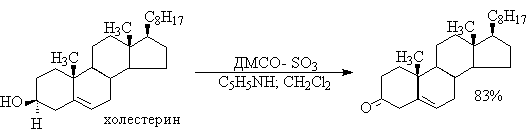
Эти методы окисления вытеснили старый громоздкий способ окисления вторичных спиртов по Оппенауэру, который заключается в нагревании спирта с алкоголятом алюминия в присутствии карбонильного соединения в качестве акцептора гидрид-ионов. Этот процесс обратим (обратная реакция называется восстановлением по Меервейну-Понндорфу-Верлею). Равновесие можно сместить вправо, если выбрать сильный акцептор гидрид-иона - п-хинон, бензофенон, хлоранил (2,3,5,6-тетрахлор-1,4-бензохинон).
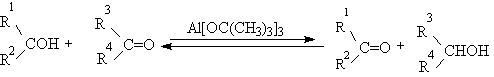
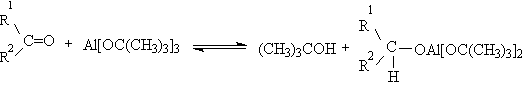
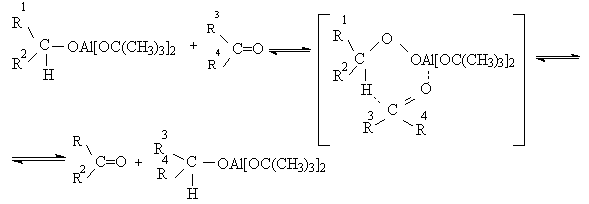
Окисление спиртов по Оппенауэру в теоретическом отношении представляет собой пример окислительного процесса с переносом гидрид-иона от восстановителя к окислителю в одну стадию, в то время, как в выше описанных процессах окисление спиртов осуществляется в несколько стадий с последовательным переносом одного или нескольких электронов.
Для получения альдегидов и кетонов из первичных и вторичных спиртов можно пользоваться реакцией каталитического дегидрирования. Так в промышленности из метанола получают формальдегид, из бутилового спирта -масляный альдегид и из циклогексанола - циклогексанон. В качестве катализатора используется медь, серебро, хромит меди.
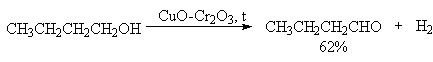
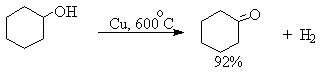
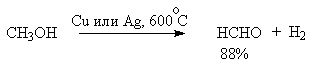
6. Защитные группы для гидроксильной группы
При осуществлении многостадийных синтезов сложных органических молекул часто необходимо защитить гидроксильную группу, чтобы провести требуемую реакцию по другой функциональной группе. Введение "защиты" включает три стадии: 1) образование инертного производного; 2) выполнение требуемого превращения с другой функциональной группой и 3) снятие защитной группы. Для гидроксильной группы хорошо зарекомендовавшим себя методом является образование ацеталя в результате кислотно-катализируемого присоединения спирта к 2,3-дигидропирану.
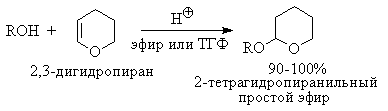
Подобно простым эфирам, тетрагидропиральные (ТГП) эфиры инертны по отношению к нуклеофильным агентам, сильным основаниям (RMgX, RLi, NaH, NaNHz, RONa и др.), а также окислителям и восстановителям. Однако ТГП-группа чувствительна к кислотному расщеплению и легко удаляется в растворе хлористого водорода в метаноле.
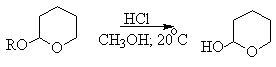
Ниже приведен типичный пример использования
защитной ТГП-группы.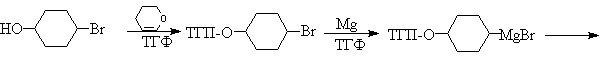
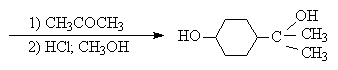
Другой защитной группой является триметилсилильная группа, которая вводится с помощью (СНз)зSiС1. Эта группа легко удаляется под действием фторид-иона.
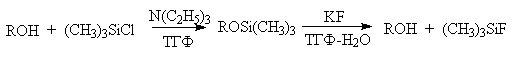
Однако триметилсилильная защитная группа малоустойчива по отношению как к кислотным, так и основным агентам и она легко снимается даже при хроматографии на силикагеле. Значительно более устойчива трет-бутилдиметилсилильная группа, которая вводится с помощью [(СНз)зС](СНз)281С1 и удаляется обработкой KF или (C4H9)4NF. Две другие защитные группы -бензильная и трифенилметильная (тритильная) вводятся с помощью бензилбромида и трифенилхлорметана в присутствии основания - третичного амина, а синаются или каталитическим гидрогенолизом или действием раствора HBr в ледяной уксусной кислоте.
![]()