3.3. Алкилирование по Фриделю-Крафтсу
Реакция Ш.Фриделя-Дж.Крафтса (1877 г.)
представляет собой удобный метод прямого
введения алкильной группы в ароматическое
кольцо. Алкилирование ароматических соединений
осуществляется под действием алкилгалогенидов,
только в присутствии в качестве катализатора
подходящей кислоты Льюиса: AlBr3, AlCl3, GaBr3,
GaCl3, BF3, SbF5, SbCl5, FeCl3,
SnCl4, ZnCl2 и др. ![]()
Наиболее активными катализаторами являются безводные сублимированные бромиды алюминия и галлия, пятифтористая сурьма, хлориды алюминия и галлия, менее активны галогениды железа (III), SbCl5, к малоактивным катализаторам относятся SnCl4 и ZnCl2. В целом активность кислот Льюиса, как катализаторов алкилирования бензола, уменьшается в ряду AlBr3> GaBr3> AlCl3> GaCl3> FeCl3> SbCl5> TiCl4> BF3> BCl3> SnCl4> SbCl3. Самым распространенным катализатором этой реакции является предварительно сублимированный хлористый алюминий.
Кинетическое изучение реакции Фриделя-Крафтса провести нелегко, поскольку скорость реакции очень чувствительна к следам влаги, реакционная смесь расслаивается в негомогенные слои, первоначальные продукты реакции легко изомеризуются и, кроме того, кинетика сильно зависит от растворителя. Несмотря на эти трудности, основные закономерности этой реакции в настоящее время достаточно хорошо выяснены. Например, бензилирование хлористым бензилом в нитробензоле в присутствии безводного AlCl3 в качестве катализатора описывается кинетическим уравнением третьего порядка v=k[RHal][ArH][AlCl3]. Поэтому механизм реакции можно представить следующей схемой:
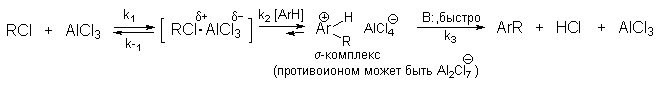
где В: =AlCl4- ; H2O или другое основание. Скорость реакции лимитируется второй стадией
Точное строение интермедиата (RCl .AlCl3) неизвестно. В принципе, можно представить целый ряд структур от молекулярного комплекса до диссоциированных карбокатионов.
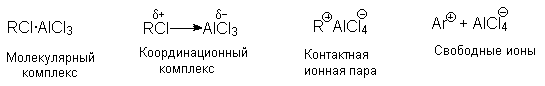
Участие свободных карбокатионов как алкилирующих агентов маловероятно.
Если бы алкилирующими агентами были свободные карбокатионы, то медленной стадией была бы стадия их образования (k1), а реакция с аренами была бы быстрой и третьего порядка не должно было наблюдаться. Крайне маловероятно, что алкилирующим агентом является молекулярный комплекс. При низких температурах иногда удается выделить комплексы алкилгалогенидов с кислотами Льюиса. Для них характерен медленный обмен галогенов по схеме:
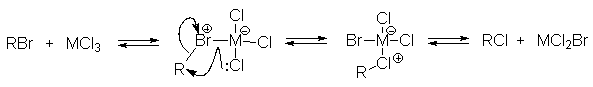
Скорость обмена возрастает в ряду перв.R< втор.R<трет.R, что можно объяснить и ион-парным строением, и структурой координационного аддукта.
Многие исследователи, работающие в данной области, полагают, что строение комплексов RX. MXn постепенно изменяется от структуры координационного аддукта в случае R=СН3 до структуры ионной пары в случае R=t-Bu, однако экспериментально это пока не подтвердено.
Способность атома галогена в RX к комплексообразованию с AlCl3 или другой жесткой кислотой Льюиса резко уменьшается от фтора к иоду, вследствие этого активность алкилгалогенидов в качестве алкилирующих агентов в реакции Фриделя-Крафтса также уменьшается в ряду RF> RCl> RBr> RI. По этой причине алкилиодиды не применяют в качестве алкилирующего агента. Различие в активности алкилфторидов и алкилбромидов настолько велико, что позволяет селективно замещать фтор в присутствии брома в одной и той же молекуле.
3.3.2. Реакция алкилирования ароматических углеводородов в органическом синтезе
Ароматические углеводороды легко алкилируются под действием самых разнообразных алкилгалогенидов, аллилгалогенидов, бензилгалогенидов и триарилметилгалогенидов в присутствии AlCl3 или AlBr3, а также FeCl3 или FeBr3 при 0-25оС или при более высокой температуре. Реакционная способность уменьшается в ряду (C6H5)3CX> (C6H5)2CHX> C6H5CH2X> CH2=CHCH2X> R3CX> R2CHX> RCH2X> CH3X.
Алкилирование аренов по Фриделю-Крафтсу как синтетический метод имеет три серьезных недостатка, ограничивающих его применение в органическом синтезе. Один из них заключается в том, что первоначально образующийся продукт алкилирования более реакционноспособен, чем исходный арен. Поэтому алкилирование аренов алкилгалогенидами при соотношении реагентов, близком к эквимольному, приводит к образованию значительного количества продуктов полиалкилирования. В этом отношении алкилирование сильно отличается от нитрования и галогенирования. Для того, чтобы свести полиалкилирование к минимуму, используют большой избыток ароматического углеводорода. В этом случае он выполняет роль и реагента, и растворителя.
Другой недостаток метода алкилирования по Фриделю-Крафтсу связан с изомеризацией алкилирующего агента в ходе реакции, в результате чего образуется смесь изомерных продуктов алкилирования. Классическим примером является алкилирование бензола н-пропилхлоридом, где получается смесь н-пропил- и изопропилбензолов, в которой изомеризованный продукт оказывается доминирующим.
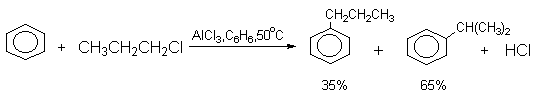
Изопропилбензол получается в результате изомеризации 1-хлорпропана в 2-хлорпропан под действием кислоты Льюиса.
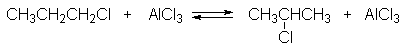
Вторичные алкилгалогениды в реакции алкилирования ароматических углеводородов более реакционноспособны, чем первичные, поэтому доля изомеризованного продукта оказывается выше, чем алкилбензола с первичной алкильной группой.
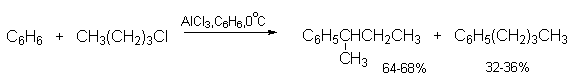
Таким образом, из первичных алкилгалогенидов образуется смесь первичных и вторичных алкилбензолов. По этой причине н-алкил-бензолы целесообразно получать не алкилированием, а ацилированием аренов по Фриделю-Крафтсу с последующим восстановлением жирно-ароматических кетонов по Клемменсену.
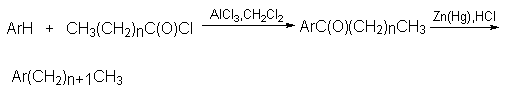
При алкилировании бензола неопентилхлоридом или изобутилхлоридом получается только трет-алкилбензол.
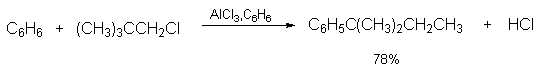
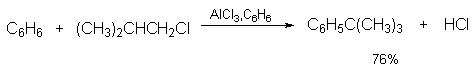
Третье ограничение, препятствуещее применению реакции алкилирования по Фриделю-Крафтсу, связано с миграцией алкильных групп в конечном продукте реакции. Алкилирование толуола 2-хлорпропаном и AlCl3 в ацетонитриле при 0оС приводит к смеси 63% орто-цимола (орто-изопропилтолуола), 25% пара-цимола и 12% мета-цимола. Однако уже при 25оС в присутствии AlCl3 (2 моля) и HCl образуется только мета-изомер цимола. При растворении смеси трех изомерных цимолов в смеси безводной HF и BF3 уже через10 минут образуется чистый мета-цимол. Аналогичный результат наблюдается при алкилировании пара-ксилола 2-хлорпропаном, где наряду с ожидаемым продуктом 2,5-диметилкумолом получается и 3,5-диметилкумол.
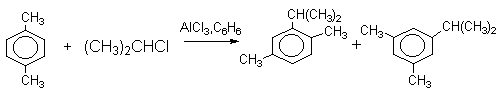
Алкилирование по Фриделю-Крафтсу относится к немногочисленной группе обратимых реакций электрофильного ароматического замещения, подчиняющихся термодинамическому контролю, когда в продуктах реакции преобладает более стабильные 1,3-диалкил- или 1,3,5,-триалкилбензолы.
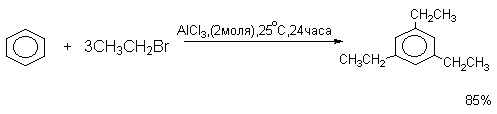
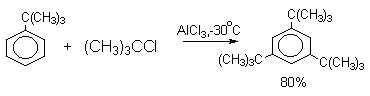
Изомеризация первоначально образующихся продуктов алкилирования в присутствии галогеноводорода и кислоты Льюиса происходит на стадии образования аренониевого иона за счет внутримолекулярного 1,2-сдвига алкильной группы. Для взаимных превращений пара-, мета- и орто-ксилолов изомеризация описывается следующим образом:
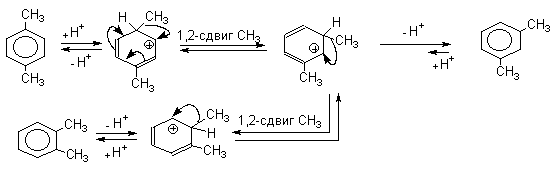
В результате перемещения метильной группы в аренониевых ионах между тремя изомерами ксилола устанавливается равновесие, в котором всегда преобладает наиболее стабильный мета-ксилол. В зависимости от температуры в смеси содержится 52-60% мета-ксилола, 23-24% пара-ксилола и 16-25% орто-ксилола. В жестких условиях изомеризация алкилбензолов приобретает межмолекулярный характер, в результате чего из ксилолов образуется смесь, содержащая три-, тетра- и пентаметилбензолы наряду с эквивалентными количествами толуола и бензола.
Ароматические соединения, содержащие электроноакцепторные заместители NO2, NO,CN,COOR и др., не алкилируются в условиях реакции Фриделя-Крафтса. Ароматические амины и фенолы связывают кислоты Льюиса в нереакционноспособный донорно-акцепторный комплекс, где неподеленная пара электронов кислорода или азота координируется с атомом металла кислоты Льюиса. Поэтому для алкилирования этих соединений в ароматическое ядро используют другие методы.
Для алкилирования ароматических углеводородов вместо алкилгалогенидов можно использовать спирты; в качестве катализаторов в этом случае берут BF3, фосфорную, полифосфорную или серную кислоты.
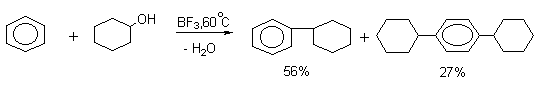
Катализатор следует применять в стехиометрическом количестве, так как вода, образующаяся в результате реакции, связывает BF3 или другой кислотный агент. Недостатки этого метода алкилирования те же, что и при алкилировании с помощью алкилгалогенидов.
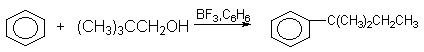
Два изомерных спирта - пентанол-2 и пентанол-3 при взаимодействии с бензолом в присутствии BF3 образуют смесь 2-фенилбутана и 3-фенилбутана в одном и том же соотношении 2:1.
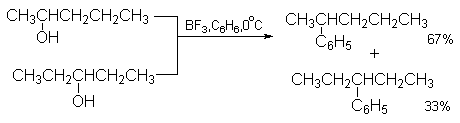
Оба катиона (или донорно-акцепторных комплекса), образующихся из изомерных спиртов, являются вторичными и близки по стабильности, поэтому соотношение продуктов алкилирования 2:1 отражает статистическую предпочтительность 2-пентилкатиона.
Алкилирование можно проводить и внутримолекулярно, что составляет одно из наиболее важных синтетических приложений этой реакции. Циклизация происходит только в том случае, когда образуется новый шести- или пятичленный цикл.
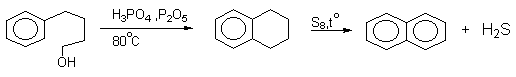
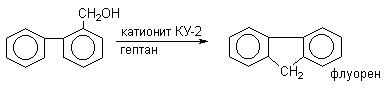
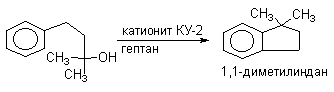
Наиболее дешевыми реагентами для алкилирования аренов в промышленном масштабе являются алкены - этилен, пропилен, изобутилен и др. Эти реакции лежат в основе крупнотоннажного производства этилбензола, кумола. Типичными катализаторами таких процессов служат системы HCl - AlCl3, или HF - BF3; H3PO4, HF.
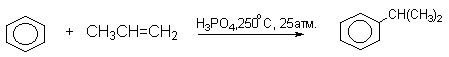
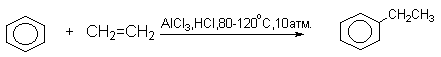
Наряду с моноалкилбензолами всегда образуются продукты диалкилирования.
Кроме спиртов и алкенов для алкилирования ароматических систем довольно широко используется формальдегид. Так в присутствии минеральных кислот реакция бензола с формальдегидом приводит к дифенилметану с высоким выходом (при использовании 85%-ной серной кислоты выход составляет 80%).
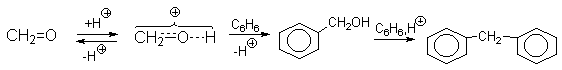
Если реакцию алкилирования ароматических соединений формальдегидом проводить в присутствии избытка газообразного хлористого водорода, то ее результатом будет образование соответствующих хлорметильных производных (реакция хлорметилирования).
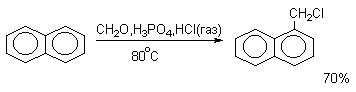
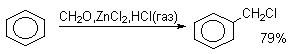
Толуол и анизол хлорметилируются в системе CH2O-ZnCl2-HCl исключительно в пара-положение. Реакция хлорметилирования протекает через первоначальное образование гидроксиметильных производных, которые в присутствии избытка HCl (газ) превращаются в хлорметильные производные.